|
Help
1. Summary
 |
|
|
Frag1D is a web server for predicting one-dimensional (1D)
structure of proteins from amino acid sequences. It predicts
three types of 1D structures, i.e. three-state secondary
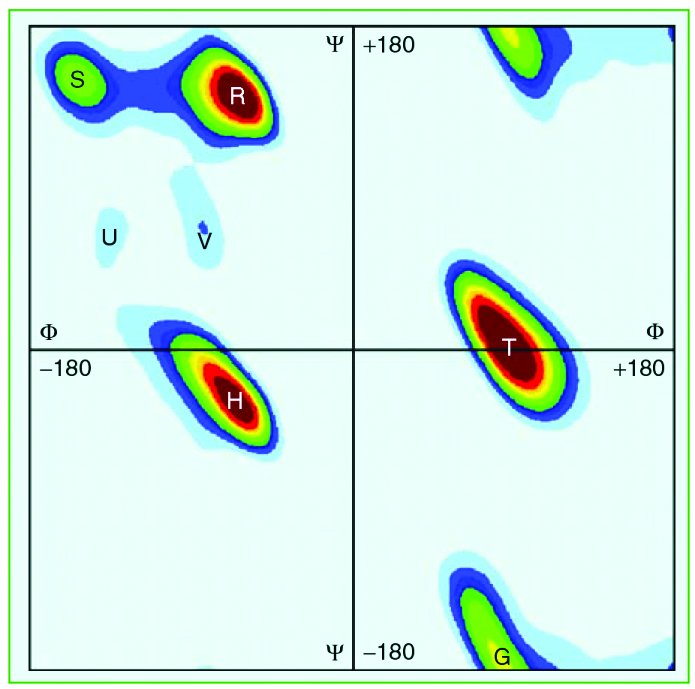
structure (H: Helix, S: Sheet, R: Random Coil), eight-state
Shape Strings (R, S, U, V, A, K, G and T, see the definition in
Figure 1) and three-state Shape Strings (R, S, U and V to S; A
and K to H; G and T to T).
Background: The precise prediction of one-dimensional (1D) protein structure
as represented by the protein secondary structure and 1D string of
discrete state of dihedral angles (i.e. Shape Strings) is a
prerequisite for the successful prediction of three-dimensional
(3D) structure as well as protein-protein interaction. We have
developed a novel 1D structure prediction method, called Frag1D,
based on a straightforward fragment matching algorithm and
demonstrated its success in the prediction of three sets of 1D
structural alphabets, i.e. the classical three-state secondary
structure, three- and eight-state Shape Strings.
By exploiting the vast protein sequence and protein
structure data available, we have brought secondary-structure
prediction closer to the expected theoretical limit. When tested
by a leave-one-out cross validation on a non-redundant set of PDB
cutting at 30% sequence identity containing 5860 protein chains,
the overall per-residue accuracy for secondary-structure
prediction, i.e. Q3 is 82.9%. The overall per-residue accuracy for
three- and eight-state Shape Strings are 85.1 and 71.5%,
respectively. We have also benchmarked our program with the latest
version of PSIPRED for secondary structure prediction and our
program predicted 0.3% better in Q3 when tested on 2241 chains
with the same training set. For Shape Strings, we compared our
method with a recently published method with the same dataset and
definition as used by that method. Our program predicted at 2.2%
better in accuracy for three-state Shape Strings. By
quantitatively investigating the effect of data base size on 1D
structure prediction we show that the accuracy increases by 1%
with every doubling of the database size.
|
3. Usage
|
|
|
Input to the server is one or several amino acid sequences (up to
) in FASTA format. The user can either paste
one or more sequences in the text-area provided, or, alternatively,
upload a file in ASCII format.
Example input:
>2az4_A mol:protein length:429 hypothetical protein EF2904
MESKAKTTVTFHSGILTIGGTVIEVAYKDAHIFFDFGTEFRPELDLPDDHIETLINNRLVPELKD
LYDPRLGYEYHGAEDKDYQHTAVFLSHAHLDHSRMINYLDPAVPLYTLKETKMILNSLNRKGDFL
IPSPFEEKNFTREMIGLNKNDVIKVGEISVEIVPVDHDAYGASALLIRTPDHFITYTGDLRLHGH
NREETLAFCEKAKHTELLMMEGVSISFPEREPDPAQIAVVSEEDLVQHLVRLELENPNRQITFNG
YPANVERFAKIIEKSPRTVVLEANMAALLLEVFGIEVRYYYAESGKIPELNPALEIPYDTLLKDK
TDYLWQVVNQFDNLQEGSLYIHSDAQPLGDFDPQYRVFLDLLAKKDITFVRLACSGHAIPEDLDK
IIALIEPQVLVPIHTLKPEKLENPYGERILPERGEQIVL
|
4. Output
|
|
The result file has eight columns, they are
Num, AA, Sec, ConfSec, S8, ConfS8, S3 and ConfS3.
Explanation:
Num residue index in the sequence.
AA one-letter amino acid code.
Sec Predicted three state secondary structure,
(H, S and R).
ConfSec Confidence of the predicted secondary structure.
S8 Predicted 8 state Shape String,
(R, S, U, V, A, K, G and T).
ConfS8 Confidence of the predicted 8 state Shape String.
S3 Predicted 3 state Shape String
(R, S, U, V -> S; A, K -> H; G, T -> T).
ConfS3 Confidence of the predicted 3 state Shape String.
|
The output for the example sequence can be found here
|
5. References
|
|
Frag1D: [Please cite this paper if you find PredZinc useful in your research]
Tuping Zhou*, Nanjiang Shu* and Sven Hovmöller. A Novel Method for Accurate One-dimensional Protein Structure Prediction based on Fragment Matching, Bioinformatics, 2010;26(4):470-477. (*Co-first author)
[PubMed]
|
5. Contact
|
|
|
Nanjiang Shu
Department for Biochemistry and Biophysics
The Arrhenius Laboratories for Natural Sciences
Stockholm University
SE-106 91 Stockholm, Sweden
Science for Life Laboratory
Box 1031, 17121 Solna, Sweden
|
|
|
|